News Message
AI 分子动力学:MDtrajNet-1
- by wittx 2025-06-27
目录
即便顶尖 AI 模型,在精确理解和操作分子语言方面也远未达标,MolLangBench 揭示了这一严峻现实。
SGPO 框架用少量实验数据就能高效指导生成模型优化蛋白质,完胜传统方法。
TxPert,这款深度学习框架,凭借生物知识图谱,能精准预测基因扰动下的转录组反应,尤其擅长应对未知扰动或细胞类型等“分布外”挑战。
PepINVENT 能设计包含天然及非天然氨基酸的新型肽,极大拓展了药物研发的化学空间与可能性。
MDtrajNet-1 以其直接预测原子轨迹的能力,将分子动力学模拟的速度和效率提升到了新高度,同时保持了高精度。
1. AI 搞不定化学语言?MolLangBench 拷问 SOTA 模型!
AI 在化学领域的语言理解能力究竟如何?MolLangBench 这个新出炉的基准测试,给出了相当硬核的答案。它不是简单地跑跑分,而是系统性地考察 AI 模型在三大关键分子语言任务上的表现:一是结构识别,模型得能从 SMILES 字符串或分子图像中精准提取原子邻居、化学键类型、环连接点、官能团等详细信息;二是语言指导的分子编辑,比如根据“替换某个基团”或“稠合两个环”这类简洁的人话指令,对分子进行修改;三是自然语言生成分子,模型需要根据包含立体化学和位置细节的丰富文本描述,从无到有构建出完整的分子结构。
这个基准测试的一大特点就是“严谨”。所有任务都设计得让输出结果具有唯一性,避免模棱两可。而且,产出的数据都经过化学专家的多轮细致标注和验证,确保了数据的“含金量”,力求贴近真实世界中化学家进行推理时的复杂需求。
MolLangBench 支持多种输入模式,包括 SMILES 化学式、二维分子图像,未来或许还能加上分子图。不过,研究者提到,图语言模型目前发展还不够充分,所以这次的评估没有包含它们。
那么,AI 模型的实际表现怎么样呢?结果可能让不少人有点意外。即使是像 GPT-4o 这样的顶尖大语言模型,在结构识别任务上的准确率也只有 79.2%,而在更具挑战性的分子生成任务上,准确率更是跌到了 29.0%。至于视觉语言模型,比如 GPT Image 1,在生成任务上基本是“交了白卷”。
研究者发现,模型常见的“翻车点”主要集中在几个方面:无法准确找到特定原子、搞不清分子的立体化学构型、生成的 SMILES 分子式经常无效。另外,大语言模型本身的分词(tokenization)问题,也常常导致它们在枚举原子数量或者操作分子结构时出错。
构建这样一个高质量的化学基准测试,工作量可不小。据研究者透露,单单是编辑和生成任务的标注,平均每个样本就要耗费 40 到 60 分钟,可见其复杂和精细程度。为了未来能有更多数据,研究团队也提出,可以尝试通过基于规则的方法来合成更多分子描述文本。
MolLangBench 为评估 AI 在化学结构理解和操作方面的能力设定了一个新标杆。它不仅清晰地暴露了当前大语言模型和视觉语言模型在该领域的局限性,也为后续开发出更懂化学、更智能的 AI 系统奠定了坚实的基础。
📜Paper: https://arxiv.org/abs/2505.15054
2. AI 蛋白设计新利器 SGPO
蛋白设计又出新招!研究者们整了个新框架叫 SGPO (Steered Generation for Protein Optimization)。简单说,它能用上百来条真实的蛋白质功能实验数据,去“指导”那些已经在大规模自然序列上训练过的生成模型(比如离散扩散模型或自回归语言模型)。这样一来,模型就能更聪明地探索和优化蛋白质序列,比传统的微调或者零样本生成方法效率高多了。
研究团队在 TrpB 和 CreiLOV 这两种基准蛋白质上,全面评估了多种生成模型(连续扩散、不同噪声类型的离散扩散、自回归预训练语言模型)和多种指导策略(比如分类器指导 CG、DAPS 和 NOS)。结果发现,哪怕只有 200 条标记序列,SGPO 也能表现出色,尤其是在数据量少的时候,效果甩开 DPO 这类监督微调方法好几条街。特别点赞的是,采用均匀噪声的离散扩散模型 (D3PM) 配上 DAPS 或 CG 指导策略,总能生成功能又强、多样性又好的蛋白质序列。相比之下,通过 DPO 微调语言模型就有点拉胯,而且对超参数还特别敏感。
SGPO 还能玩转类似贝叶斯优化的自适应优化流程。具体怎么操作呢?研究者们用了一套集成的适应度预测器来指导生成过程,有点像 Thompson 抽样。通过这种迭代操作,能找到功能更强的蛋白质。
而且,SGPO 的即插即用指导方式,计算资源需求很小。预训练模型速度快,实际“指导”过程也就几分钟的事儿。跟那些基于强化学习或者需要彻底微调的方法比起来,简直不要太省心,后者通常需要更长的训练和调优周期。这套框架最大的亮点就是灵活:生成先验模型预训练一次就能反复用,而像 DAPS 或 CG 这样的指导策略,可以直接叠加上去,根本不用动模型的权重。
SGPO 成功避开了其他方法的坑:它能处理更大的序列空间 (N > 9),既利用了进化先验知识,也用上了实验数据,还不会像传统定向进化那样陷入局部搜索的低效怪圈。
通过对指导策略和模型架构的系统分析,这项工作不仅给出了实用建议,还贡献了一个强大的开源工具。这为现实世界的蛋白质工程树立了新标杆,可以说是为 ML 辅助蛋白质设计领域注入了新活力。
💻Code: https://github.com/jsunn-y/SGPO
📜Paper: https://arxiv.org/abs/2505.15093
3. TxPert:AI 精准预测未知基因扰动
细胞对基因扰动的反应,一直是生物学研究的重点和难点。现在,一款名为 TxPert 的深度学习框架横空出世,它能精准预测细胞在基因扰动下的转录组反应,厉害的是,即便是在全新的扰动条件或之前未曾见过的细胞类型(即“分布外”场景),TxPert 依旧能给出靠谱的预测。
TxPert 的“超能力”很大程度上归功于它对生物知识图谱的巧妙运用。研究者将精心整理的基因间相互作用网络,比如大家熟知的 STRINGdb、GO(基因本体论)、PxMap 和 TxMap,都整合进了模型。具体来说,TxPert 的架构包含两大核心模块:一个是基础状态编码器,负责捕捉细胞自身的“上下文”信息;另一个则是扰动编码器,它基于图神经网络(GNN)构建,专门用来学习和编码这些生物知识图谱中蕴含的复杂关系。
模型的工作方式也颇具新意,研究者称之为“潜空间迁移”方法。简单讲,就是先把细胞在未受扰动时的“平静”状态(对照组)嵌入到一个数学空间里,然后,再把从生物知识图谱中学到的扰动效应(以向量形式表示)“叠加”上去,这样就能推测出细胞在受到扰动后的转录组图谱了。这种设计的好处在于,它让模型可以模块化地、组合式地对多种扰动进行推理。
实践是检验真理的唯一标准。在多项硬核的“分布外”预测任务中,TxPert 的表现确实亮眼,无论是预测单一未知扰动、还是更复杂的双重未知扰动,亦或是在全新的细胞系中进行预测,它都全面超越了像 GEARS 和 scLAMBDA 这样的顶尖模型。在某些情况下,TxPert 的预测精准度甚至快要赶上真实实验的可重复性水平了!特别是在最具挑战性的“零样本”预测任务——即在模型从未“见过”的细胞系中进行预测时,TxPert 依然能够凭借其对先验知识的整合以及对对照组变异性的精细建模,大幅超越其他基线模型。这无疑为它在真实世界的药物发现流程中进行迁移学习带来了曙光。
研究者通过消融实验进一步证实,生物知识图谱的质量和结构对模型性能至关重要。而且,将多个不同来源的知识图谱(比如 STRINGdb + PxMap + GO + TxMap)结合起来使用,还能给模型的预测准确性再上一个“buff”。为了应对真实世界中知识图谱不可避免的噪声和不完整性,TxPert 还装备了基于注意力机制的 GNN 和图 Transformer(如 GAT、GAT-Hybrid 和 Exphormer-MG),这使得模型能够跨多个知识来源进行稳健推理,并捕捉图中远距离的依赖关系。
除了模型本身,TxPert 的评估框架也是一大创新。它没有停留在传统的衡量标准上,而是更强调那些能够衡量模型区分不同生物学扰动能力的指标(如 Pearson ∆ 和检索指标),这与真实实验的目标更为契合。在某些扰动场景下,TxPert 的表现已经接近人类专家的水平,这意味着它有潜力成为昂贵实验室操作的可靠“数字替身”,为扰动生物学领域的虚拟筛选、药物靶点优先级排序和假设检验打开了新大门。
通过将精心策划的生物学先验知识、大规模扰动数据与现代 GNN 架构深度融合,TxPert 为模拟细胞应答机制打下了一个坚实且可扩展的基础。
📜Paper: https://arxiv.org/abs/2505.14919
💻Code: https://github.com/valence-labs/TxPert
4. PepINVENT: AI 驱动非天然肽设计,解锁新药!
想让肽类药物设计跳出那 20 种天然氨基酸的“舒适圈”吗?现在,机会来了!研究者们带来了一款名为 PepINVENT 的生成式 AI 框架,它简直是肽设计领域的“变形金刚”。这套系统基于 REINVENT 平台扩展而来,最亮眼的地方在于,它不仅能玩转天然氨基酸,更能将超过一万种可合成的非天然氨基酸(NNAAs)纳入囊中,化学多样性直接拉满!
PepINVENT 的学习方式也挺有意思,它采用了一种氨基酸水平的掩码填充策略。简单说,就是给它一个“残缺”的肽序列,让它学习如何填补空缺,同时保证整个分子的化学结构没毛病。这背后的一大功臣是 CHUCKLES,一种在单体层面化学标准化的 SMILES 表示法。有了它,PepINVENT 就能在原子级别进行精准控制,确保肽序列的有效拼接,无论是线性肽、头尾相连的环肽、二硫键桥接肽,还是侧链到尾部的环肽,它都能轻松驾驭,并且成功学习到拓扑结构的约束。生成的肽序列不仅有效性超高(98-100%),而且在各种拓扑结构中都展现出极高的独特性。
PepINVENT 还能“脑洞大开”,生成训练集中从未见过的全新氨基酸结构,目前已在采样中鉴定出超过 9 万种。研究发现,采用多项式采样能比束搜索(beam search)产生更广泛的多样性,充分显示了 PepINVENT 在探索未知化学空间方面的强大实力。
当然,光会设计还不够,得能“按需定制”。PepINVENT 引入了强化学习机制,摇身一变成为目标导向的设计高手。它可以针对性地优化肽的拓扑特征(比如环的大小),或者像溶解度、细胞渗透性这样的关键理化性质,实现多参数优化 (MPO)。举个例子,在一个实际应用中,研究者们用 PepINVENT 重新设计了一种已知的巨噬细胞 Rev 结合肽 (RBP),通过学习到的评分函数和强化学习引导的采样,成功提升了其溶解度和细胞渗透性,大大增强了其治疗潜力。整个优化过程,通常在不到 50 个强化学习步骤内就能搞定。PepINVENT 还支持集成 CAMSOL-PTM 和渗透性分类器等属性预测工具,让生物学特性的精细调控成为可能。
PepINVENT 为肽生成模型树立了新的标杆,它提供了精细、灵活且可扩展的新型肽设计方案,在模拟肽、先导化合物优化和治疗药物开发等领域都有着广阔的应用前景。
📜Paper: https://doi.org/10.1039/d4sc07642g
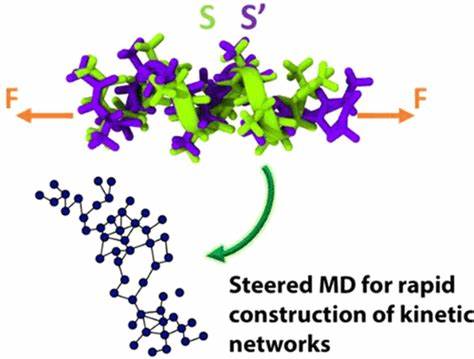
5. AI 预测分子动力学:MDtrajNet-1 提速百倍!
AI 在分子动力学领域又整了个活儿!隆重介绍 MDtrajNet-1,这个模型直接革了传统分子动力学(MD)的命。它不再一步步吭哧吭哧地算力、积分,而是选择了一条捷径——直接预测原子在 4D 时空中的完整轨迹。
结果怎么样?模拟速度直接起飞,比传统 MD 快了不止 100 倍,就算是那些用了机器学习力场(MLFF)加速的传统方法,也得靠边站。
这款模型的核心是个基于 Transformer 的 E(3) 等变神经网络。它能聪明地学习原子位置、速度、元素类型和时间间隔之间的复杂关系,然后“一步到位”给出未来的分子构型。这种设计不仅让大规模并行计算成为现实,效率大大提升,而且精度也一点没落下。
说到精度,MDtrajNet-1 相当给力。在 10 飞秒这样的短时间预测里,它的误差能控制在亚皮米(sub-picometer)级别,这水平,直接叫板从头算分子动力学(ab initio MD)。要是看 10 皮秒的长时间预测,它还原光谱特征的保真度,跟 DFT(密度泛函理论)级别的模拟有一拼。
更牛的是,达到这种效果,它用的训练数据其实只是 ANI-1xMD 数据集里的一小部分——从包含 173 个分子体系(最多 9 个原子)的 100 万个数据点里学出来的。尽管训练数据在规模和多样性上都比较“经济”,MDtrajNet-1 却展现出了惊人的泛化能力,对于之前没“见过”的分子和化学空间,也能很好地应对。
跟前辈 GICnet 模型比起来,MDtrajNet-1 在精度、可扩展性和泛化性上都秀了一把肌肉。这主要归功于架构上的升级,比如多头等变注意力机制和对局部原子环境更精细的表征。
MDtrajNet-1 的应用场景也很多样。除了标准的 NVE 系综,研究者们发现,稍作调整(重新训练),它也能胜任 NVT 系综的模拟,即使加上了恒温器带来的额外复杂性,它给出的振动光谱质量依然杠杠的。它的能力还覆盖到了周期性体系和不同类型的相互作用。无论是模拟金刚石晶格,还是 Lennard-Jones 流体(都带周期性边界条件),它都能成功捕捉到像径向分布函数这样的关键结构特征。
更让人惊喜的是它的迁移学习本领。在一个包含 22 个原子的丙氨酸二肽分子上,只用很短的轨迹数据对 MDtrajNet-1 进行微调,它就能比那些在相同数据上训练的机器学习力场(MLIPs)更准确地复现分子长时间的构象动力学(比如拉马钱德兰图)。
从架构上看,MDtrajNet-1 采用 O(3) 等变设计,以原子为中心,计算复杂度随体系原子数线性增长。这意味着,在主流硬件(比如一块 RTX 4090 显卡)上,模拟大型体系也能保持高效。
MDtrajNet-1 的出现,无疑为开发通用的、基础性的分子模拟大模型铺平了道路。它把物理启发的架构设计和强大的生成能力巧妙地结合起来,为化学与材料科学领域实现可扩展、高精度、高效率的轨迹生成,指明了一条非常值得期待的新路子。
📜Paper: https://doi.org/10.26434/chemrxiv-2025-kc7sn
💻Code: https://github.com/dralgroup/mlatom
即便顶尖 AI 模型,在精确理解和操作分子语言方面也远未达标,MolLangBench 揭示了这一严峻现实。
SGPO 框架用少量实验数据就能高效指导生成模型优化蛋白质,完胜传统方法。
TxPert,这款深度学习框架,凭借生物知识图谱,能精准预测基因扰动下的转录组反应,尤其擅长应对未知扰动或细胞类型等“分布外”挑战。
PepINVENT 能设计包含天然及非天然氨基酸的新型肽,极大拓展了药物研发的化学空间与可能性。
MDtrajNet-1 以其直接预测原子轨迹的能力,将分子动力学模拟的速度和效率提升到了新高度,同时保持了高精度。
1. AI 搞不定化学语言?MolLangBench 拷问 SOTA 模型!
AI 在化学领域的语言理解能力究竟如何?MolLangBench 这个新出炉的基准测试,给出了相当硬核的答案。它不是简单地跑跑分,而是系统性地考察 AI 模型在三大关键分子语言任务上的表现:一是结构识别,模型得能从 SMILES 字符串或分子图像中精准提取原子邻居、化学键类型、环连接点、官能团等详细信息;二是语言指导的分子编辑,比如根据“替换某个基团”或“稠合两个环”这类简洁的人话指令,对分子进行修改;三是自然语言生成分子,模型需要根据包含立体化学和位置细节的丰富文本描述,从无到有构建出完整的分子结构。
这个基准测试的一大特点就是“严谨”。所有任务都设计得让输出结果具有唯一性,避免模棱两可。而且,产出的数据都经过化学专家的多轮细致标注和验证,确保了数据的“含金量”,力求贴近真实世界中化学家进行推理时的复杂需求。
MolLangBench 支持多种输入模式,包括 SMILES 化学式、二维分子图像,未来或许还能加上分子图。不过,研究者提到,图语言模型目前发展还不够充分,所以这次的评估没有包含它们。
那么,AI 模型的实际表现怎么样呢?结果可能让不少人有点意外。即使是像 GPT-4o 这样的顶尖大语言模型,在结构识别任务上的准确率也只有 79.2%,而在更具挑战性的分子生成任务上,准确率更是跌到了 29.0%。至于视觉语言模型,比如 GPT Image 1,在生成任务上基本是“交了白卷”。
研究者发现,模型常见的“翻车点”主要集中在几个方面:无法准确找到特定原子、搞不清分子的立体化学构型、生成的 SMILES 分子式经常无效。另外,大语言模型本身的分词(tokenization)问题,也常常导致它们在枚举原子数量或者操作分子结构时出错。
构建这样一个高质量的化学基准测试,工作量可不小。据研究者透露,单单是编辑和生成任务的标注,平均每个样本就要耗费 40 到 60 分钟,可见其复杂和精细程度。为了未来能有更多数据,研究团队也提出,可以尝试通过基于规则的方法来合成更多分子描述文本。
MolLangBench 为评估 AI 在化学结构理解和操作方面的能力设定了一个新标杆。它不仅清晰地暴露了当前大语言模型和视觉语言模型在该领域的局限性,也为后续开发出更懂化学、更智能的 AI 系统奠定了坚实的基础。
📜Paper: https://arxiv.org/abs/2505.15054
2. AI 蛋白设计新利器 SGPO
蛋白设计又出新招!研究者们整了个新框架叫 SGPO (Steered Generation for Protein Optimization)。简单说,它能用上百来条真实的蛋白质功能实验数据,去“指导”那些已经在大规模自然序列上训练过的生成模型(比如离散扩散模型或自回归语言模型)。这样一来,模型就能更聪明地探索和优化蛋白质序列,比传统的微调或者零样本生成方法效率高多了。
研究团队在 TrpB 和 CreiLOV 这两种基准蛋白质上,全面评估了多种生成模型(连续扩散、不同噪声类型的离散扩散、自回归预训练语言模型)和多种指导策略(比如分类器指导 CG、DAPS 和 NOS)。结果发现,哪怕只有 200 条标记序列,SGPO 也能表现出色,尤其是在数据量少的时候,效果甩开 DPO 这类监督微调方法好几条街。特别点赞的是,采用均匀噪声的离散扩散模型 (D3PM) 配上 DAPS 或 CG 指导策略,总能生成功能又强、多样性又好的蛋白质序列。相比之下,通过 DPO 微调语言模型就有点拉胯,而且对超参数还特别敏感。
SGPO 还能玩转类似贝叶斯优化的自适应优化流程。具体怎么操作呢?研究者们用了一套集成的适应度预测器来指导生成过程,有点像 Thompson 抽样。通过这种迭代操作,能找到功能更强的蛋白质。
而且,SGPO 的即插即用指导方式,计算资源需求很小。预训练模型速度快,实际“指导”过程也就几分钟的事儿。跟那些基于强化学习或者需要彻底微调的方法比起来,简直不要太省心,后者通常需要更长的训练和调优周期。这套框架最大的亮点就是灵活:生成先验模型预训练一次就能反复用,而像 DAPS 或 CG 这样的指导策略,可以直接叠加上去,根本不用动模型的权重。
SGPO 成功避开了其他方法的坑:它能处理更大的序列空间 (N > 9),既利用了进化先验知识,也用上了实验数据,还不会像传统定向进化那样陷入局部搜索的低效怪圈。
通过对指导策略和模型架构的系统分析,这项工作不仅给出了实用建议,还贡献了一个强大的开源工具。这为现实世界的蛋白质工程树立了新标杆,可以说是为 ML 辅助蛋白质设计领域注入了新活力。
💻Code: https://github.com/jsunn-y/SGPO
📜Paper: https://arxiv.org/abs/2505.15093
3. TxPert:AI 精准预测未知基因扰动
细胞对基因扰动的反应,一直是生物学研究的重点和难点。现在,一款名为 TxPert 的深度学习框架横空出世,它能精准预测细胞在基因扰动下的转录组反应,厉害的是,即便是在全新的扰动条件或之前未曾见过的细胞类型(即“分布外”场景),TxPert 依旧能给出靠谱的预测。
TxPert 的“超能力”很大程度上归功于它对生物知识图谱的巧妙运用。研究者将精心整理的基因间相互作用网络,比如大家熟知的 STRINGdb、GO(基因本体论)、PxMap 和 TxMap,都整合进了模型。具体来说,TxPert 的架构包含两大核心模块:一个是基础状态编码器,负责捕捉细胞自身的“上下文”信息;另一个则是扰动编码器,它基于图神经网络(GNN)构建,专门用来学习和编码这些生物知识图谱中蕴含的复杂关系。
模型的工作方式也颇具新意,研究者称之为“潜空间迁移”方法。简单讲,就是先把细胞在未受扰动时的“平静”状态(对照组)嵌入到一个数学空间里,然后,再把从生物知识图谱中学到的扰动效应(以向量形式表示)“叠加”上去,这样就能推测出细胞在受到扰动后的转录组图谱了。这种设计的好处在于,它让模型可以模块化地、组合式地对多种扰动进行推理。
实践是检验真理的唯一标准。在多项硬核的“分布外”预测任务中,TxPert 的表现确实亮眼,无论是预测单一未知扰动、还是更复杂的双重未知扰动,亦或是在全新的细胞系中进行预测,它都全面超越了像 GEARS 和 scLAMBDA 这样的顶尖模型。在某些情况下,TxPert 的预测精准度甚至快要赶上真实实验的可重复性水平了!特别是在最具挑战性的“零样本”预测任务——即在模型从未“见过”的细胞系中进行预测时,TxPert 依然能够凭借其对先验知识的整合以及对对照组变异性的精细建模,大幅超越其他基线模型。这无疑为它在真实世界的药物发现流程中进行迁移学习带来了曙光。
研究者通过消融实验进一步证实,生物知识图谱的质量和结构对模型性能至关重要。而且,将多个不同来源的知识图谱(比如 STRINGdb + PxMap + GO + TxMap)结合起来使用,还能给模型的预测准确性再上一个“buff”。为了应对真实世界中知识图谱不可避免的噪声和不完整性,TxPert 还装备了基于注意力机制的 GNN 和图 Transformer(如 GAT、GAT-Hybrid 和 Exphormer-MG),这使得模型能够跨多个知识来源进行稳健推理,并捕捉图中远距离的依赖关系。
除了模型本身,TxPert 的评估框架也是一大创新。它没有停留在传统的衡量标准上,而是更强调那些能够衡量模型区分不同生物学扰动能力的指标(如 Pearson ∆ 和检索指标),这与真实实验的目标更为契合。在某些扰动场景下,TxPert 的表现已经接近人类专家的水平,这意味着它有潜力成为昂贵实验室操作的可靠“数字替身”,为扰动生物学领域的虚拟筛选、药物靶点优先级排序和假设检验打开了新大门。
通过将精心策划的生物学先验知识、大规模扰动数据与现代 GNN 架构深度融合,TxPert 为模拟细胞应答机制打下了一个坚实且可扩展的基础。
📜Paper: https://arxiv.org/abs/2505.14919
💻Code: https://github.com/valence-labs/TxPert
4. PepINVENT: AI 驱动非天然肽设计,解锁新药!
想让肽类药物设计跳出那 20 种天然氨基酸的“舒适圈”吗?现在,机会来了!研究者们带来了一款名为 PepINVENT 的生成式 AI 框架,它简直是肽设计领域的“变形金刚”。这套系统基于 REINVENT 平台扩展而来,最亮眼的地方在于,它不仅能玩转天然氨基酸,更能将超过一万种可合成的非天然氨基酸(NNAAs)纳入囊中,化学多样性直接拉满!
PepINVENT 的学习方式也挺有意思,它采用了一种氨基酸水平的掩码填充策略。简单说,就是给它一个“残缺”的肽序列,让它学习如何填补空缺,同时保证整个分子的化学结构没毛病。这背后的一大功臣是 CHUCKLES,一种在单体层面化学标准化的 SMILES 表示法。有了它,PepINVENT 就能在原子级别进行精准控制,确保肽序列的有效拼接,无论是线性肽、头尾相连的环肽、二硫键桥接肽,还是侧链到尾部的环肽,它都能轻松驾驭,并且成功学习到拓扑结构的约束。生成的肽序列不仅有效性超高(98-100%),而且在各种拓扑结构中都展现出极高的独特性。
PepINVENT 还能“脑洞大开”,生成训练集中从未见过的全新氨基酸结构,目前已在采样中鉴定出超过 9 万种。研究发现,采用多项式采样能比束搜索(beam search)产生更广泛的多样性,充分显示了 PepINVENT 在探索未知化学空间方面的强大实力。
当然,光会设计还不够,得能“按需定制”。PepINVENT 引入了强化学习机制,摇身一变成为目标导向的设计高手。它可以针对性地优化肽的拓扑特征(比如环的大小),或者像溶解度、细胞渗透性这样的关键理化性质,实现多参数优化 (MPO)。举个例子,在一个实际应用中,研究者们用 PepINVENT 重新设计了一种已知的巨噬细胞 Rev 结合肽 (RBP),通过学习到的评分函数和强化学习引导的采样,成功提升了其溶解度和细胞渗透性,大大增强了其治疗潜力。整个优化过程,通常在不到 50 个强化学习步骤内就能搞定。PepINVENT 还支持集成 CAMSOL-PTM 和渗透性分类器等属性预测工具,让生物学特性的精细调控成为可能。
PepINVENT 为肽生成模型树立了新的标杆,它提供了精细、灵活且可扩展的新型肽设计方案,在模拟肽、先导化合物优化和治疗药物开发等领域都有着广阔的应用前景。
📜Paper: https://doi.org/10.1039/d4sc07642g
5. AI 预测分子动力学:MDtrajNet-1 提速百倍!
AI 在分子动力学领域又整了个活儿!隆重介绍 MDtrajNet-1,这个模型直接革了传统分子动力学(MD)的命。它不再一步步吭哧吭哧地算力、积分,而是选择了一条捷径——直接预测原子在 4D 时空中的完整轨迹。
结果怎么样?模拟速度直接起飞,比传统 MD 快了不止 100 倍,就算是那些用了机器学习力场(MLFF)加速的传统方法,也得靠边站。
这款模型的核心是个基于 Transformer 的 E(3) 等变神经网络。它能聪明地学习原子位置、速度、元素类型和时间间隔之间的复杂关系,然后“一步到位”给出未来的分子构型。这种设计不仅让大规模并行计算成为现实,效率大大提升,而且精度也一点没落下。
说到精度,MDtrajNet-1 相当给力。在 10 飞秒这样的短时间预测里,它的误差能控制在亚皮米(sub-picometer)级别,这水平,直接叫板从头算分子动力学(ab initio MD)。要是看 10 皮秒的长时间预测,它还原光谱特征的保真度,跟 DFT(密度泛函理论)级别的模拟有一拼。
更牛的是,达到这种效果,它用的训练数据其实只是 ANI-1xMD 数据集里的一小部分——从包含 173 个分子体系(最多 9 个原子)的 100 万个数据点里学出来的。尽管训练数据在规模和多样性上都比较“经济”,MDtrajNet-1 却展现出了惊人的泛化能力,对于之前没“见过”的分子和化学空间,也能很好地应对。
跟前辈 GICnet 模型比起来,MDtrajNet-1 在精度、可扩展性和泛化性上都秀了一把肌肉。这主要归功于架构上的升级,比如多头等变注意力机制和对局部原子环境更精细的表征。
MDtrajNet-1 的应用场景也很多样。除了标准的 NVE 系综,研究者们发现,稍作调整(重新训练),它也能胜任 NVT 系综的模拟,即使加上了恒温器带来的额外复杂性,它给出的振动光谱质量依然杠杠的。它的能力还覆盖到了周期性体系和不同类型的相互作用。无论是模拟金刚石晶格,还是 Lennard-Jones 流体(都带周期性边界条件),它都能成功捕捉到像径向分布函数这样的关键结构特征。
更让人惊喜的是它的迁移学习本领。在一个包含 22 个原子的丙氨酸二肽分子上,只用很短的轨迹数据对 MDtrajNet-1 进行微调,它就能比那些在相同数据上训练的机器学习力场(MLIPs)更准确地复现分子长时间的构象动力学(比如拉马钱德兰图)。
从架构上看,MDtrajNet-1 采用 O(3) 等变设计,以原子为中心,计算复杂度随体系原子数线性增长。这意味着,在主流硬件(比如一块 RTX 4090 显卡)上,模拟大型体系也能保持高效。
MDtrajNet-1 的出现,无疑为开发通用的、基础性的分子模拟大模型铺平了道路。它把物理启发的架构设计和强大的生成能力巧妙地结合起来,为化学与材料科学领域实现可扩展、高精度、高效率的轨迹生成,指明了一条非常值得期待的新路子。
📜Paper: https://doi.org/10.26434/chemrxiv-2025-kc7sn
💻Code: https://github.com/dralgroup/mlatom
Share Http URL: http://www.wittx.cn/get_news_message.do?new_id=1465


Best Last Month

Information industry
by wittx

Information industry
by wittx

Information industry
by wittx
Information industry
by wittx
Information industry
by wittx

Information industry
by wittx
Information industry
by wittx
Information industry
by wittx

Information industry
by wittx

Information industry
by wittx